
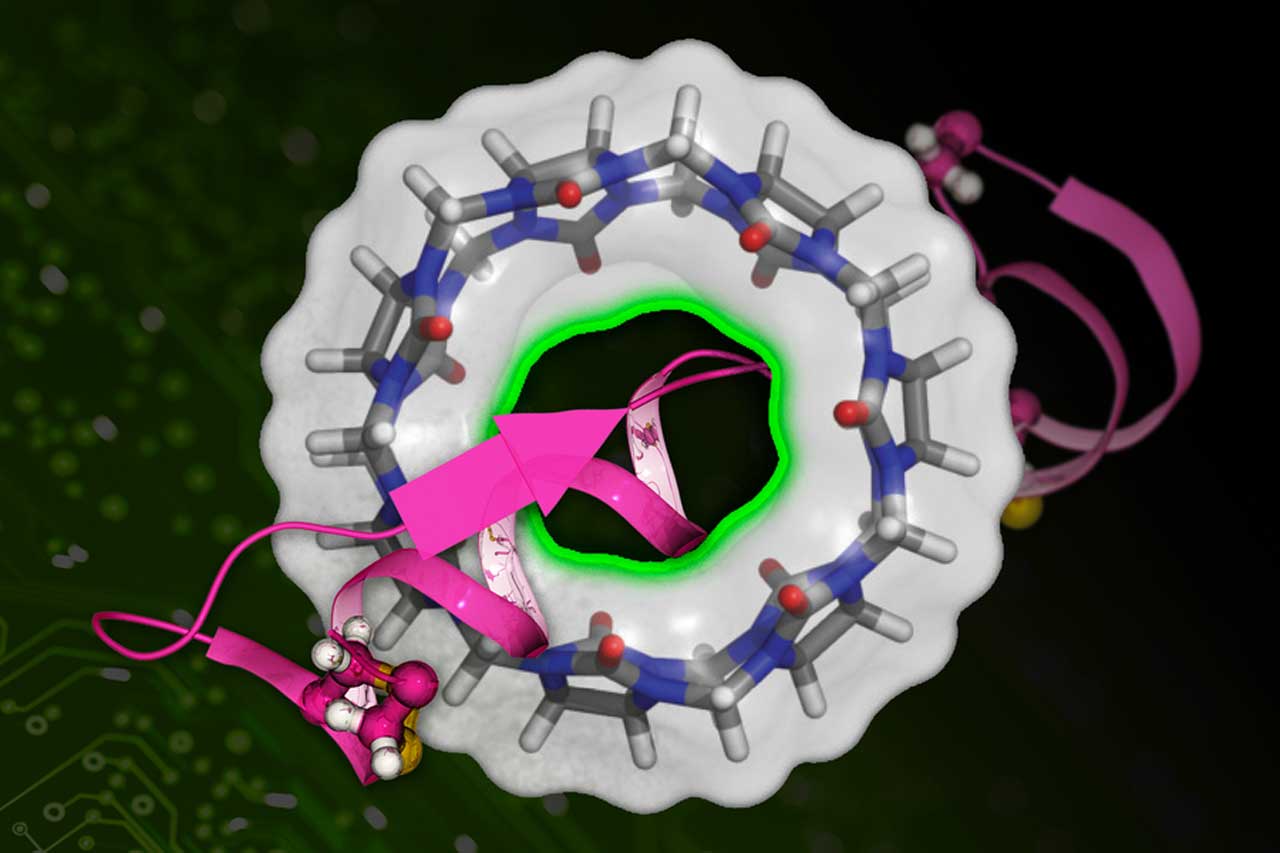
Drugs stick together by sticking to their target proteins. Although, the challenging task is to assess the stickiness.
Thanks to a new technique called DeepBAR, scientists can speedily calculate the binding affinities between drug candidates and their targets. MIT scientists develop the approach by combining chemistry and machine learning.
The DeepBAR gives accurate calculations within a short time. According to scientists, it could one day quicken the pace of drug discovery and protein engineering.
Bin Zhang, the Pfizer-Laubach Career Development Professor in Chemistry at MIT, said, “The affinity between a drug molecule and a target protein is measured by a quantity called the binding free energy — the smaller the number, the stickier the bond. A lower binding free energy means the drug can better compete against other molecules, meaning it can more effectively disrupt the protein’sprotein’s normal function. Calculating the binding free energy of a drug candidate provides an indicator of a drug’sdrug’s potential effectiveness. But it’sit’s a difficult quantity to nail down.”
DeepBAR computes binding free energy precisely, but it requires just a fraction of the calculations demanded by previous methods. The ”BAR” in DeepBar is an abbreviation of Bennett acceptance ratio. Bennett acceptance ratio is an old algorithm that is used to calculate binding free energy. Using the Bennet acceptance ratio typically requires a knowledge of two “endpoint” states (e.g., a drug molecule bound to a protein and a drug molecule completely dissociated from a protein), plus knowledge of many intermediate conditions (e.g., varying levels of partial binding), all of which bog down calculation speed.
Zhang said, “DeepBAR slashes those in-between states by deploying the Bennett acceptance ratio in machine-learning frameworks called deep generative models. These models create a reference state for each endpoint, the bound state, and the unbound state. These two reference states are similar enough that the Bennett acceptance ratio can be used directly, without all the costly intermediate steps.”
Xinqiang Ding, a postdoc in MIT’s Department of Chemistry, said, “While adapting a computer vision approach to chemistry was DeepBAR’sDeepBAR’s key innovation, the crossover also raised some challenges. These models were originally developed for 2D images. But here we have proteins and molecules — it’s a 3D structure. So, adapting those methods in our case was the biggest technical challenge we had to overcome.”
During tests using small protein-like molecules, scientists found that the DeepBAR 50 times faster than previous methods and has the same accuracy as the gold standard, but it’s much faster.
Zhang said, “With such efficiency, we can really start to think about using this to do drug screening, in particular in the context of Covid. In addition to drug screening, DeepBAR could aid protein design and engineering since the method could be used to model interactions between multiple proteins.”
In the future, the researchers plan to improve DeepBAR’sDeepBAR’s ability to run calculations for large proteins, a task made feasible by recent advances in computer science.
The research appears today in the Journal of Physical Chemistry Letters.
Continue reading DeepBAR: New technique to quickly calculate drug molecules’ binding affinity to proteins on Tech Explorist.
0 comments:
Post a Comment